Index
Indexing variants does not apply any modification to the generic pipeline. The input file format is VCF, accepting different variations like gVCF or aggregated VCFs
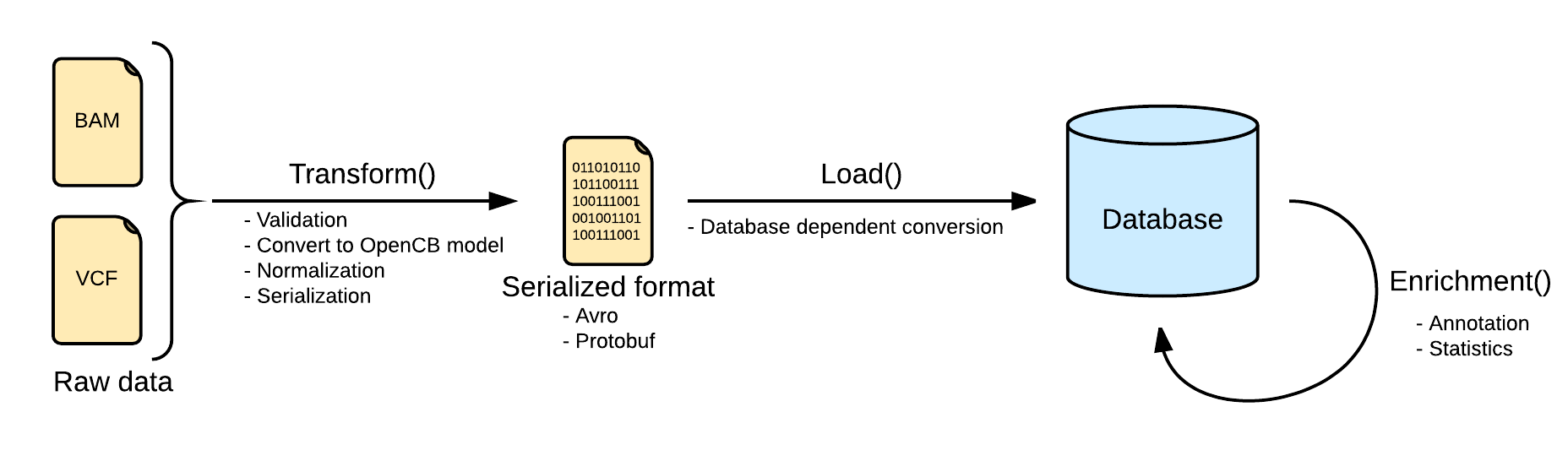
Transform
Files are converted Biodata models. The metadata and the data are serialized into two separated files. The metadata is stored into a file named <inputFileName>.file.json.gz serializing in json a single instance of the biodata model VariantSource, which mainly contains the header and some general stats. Along with this file, the real variants data is stored in a file named <inputFileName>.variants.avro.gz with a set of variant records described as the biodata model Variant.
VCF files are read using the library HTSJDK, which provides a syntactic validation of the data. Further actions on the validation will be taken, like duplicate or overlapping variants detection.
By default, malformed variants will be skipped and written into a third optional file named <inputFileName>.malformed.txt . If the transform step generates this file, a curation process should be taken to repair the file. Otherwise, the variants would be skipped.
All the variants in the transform step will be normalized as defined here: Variant Normalization. This will help to unify the variants representation, since the VCF specification allows multiple ways of referring to a variant and some ambiguities.
Load
Loading variants from multiple files into a single database will effectively merge them. In most of the scenarios, with a good normalization, merging variants is strait forward. But in some other scenarios, with multiple alternates or overlapping variants, a more complex merge is needed in order to create a consistent database. This situations can be solved when loading the file, or a posteriori in the aggregation operation.
Loading process is dependent on the implementation. Here you can see some specific information for the two implemented back-ends.
MongoDB
The MongoDB implementation stores all the variant information in one centralised collection, with some secondary helper collections. In order to merge correctly new variants with the already existing data, the engine uses a stage collection to keep track of the already loaded data. In case of loading multiple files at the same time, this files will first be written into this stage collection, and then, moved to the variants collection, all at the same time.
Using this stage collection, the engine is able to solve the complex merge situations when loading the file, without the need of an extra aggregation step. Therefore, this storage engine does not implement the aggregation operation. Depending on the level of detail required, the merge mode can be configured when loading the files.
Merge Mode
For each variant that we load we have to check if the it already exists in the database, and, in that case, merge the new data with the existing variant. Otherwise, create a new variant.
- Basic merge. Only merging variants from different sources that are the same.

For basic mode, there will be unknown values for certain positions. We can not determine if the value was missing ( ./. ), reference ( 0/0 ), or a different genotype. The output value for unknown genotypes can be modified by the user when querying. By default, the missing genotype ( ./. ) will be used.
In the advanced mode, the variants have gained a secondary alternate, and the field AD (Allele Depth) has been rearranged in order to match with the new allele order.
Performance advantages
Loading new files will be much faster with basic merge mode. Is is because we don't need now to check if the variant overlaps with any other already existing variant. We only need to know if the variant exists or not in the database, which takes a significant amount of time in advance mode.
Hadoop HBase
Tables
Limitations
- You can not load two files with the same sample in the same study. See OpenCGA#158.
There is an exception for this limitation for the scenarios where the variants were split in multiple files (by chromosome, by type, ...). In this case, you can use the parameter--load-split-data. SeeOpenCGA#696 - You can not index two files with the same name (e.g. /data/sample1/my.vcf.gz and /data/sample2/my.vcf.gz) in the same study. This limitation should not be a problem in any real scenario, where every VCF file usually has a different name. If two files have the same name, the most likely situation is that they contain the same samples, and this is already forbidden by the previous limitation.
Table of Contents: