The OpenCGA Variant Storage Engine supports basic operations to work with variant datasets.
Index
Indexing variants does not apply any modification to the generic pipeline. The input file format is VCF, accepting different variations like gVCF or aggregated VCFs
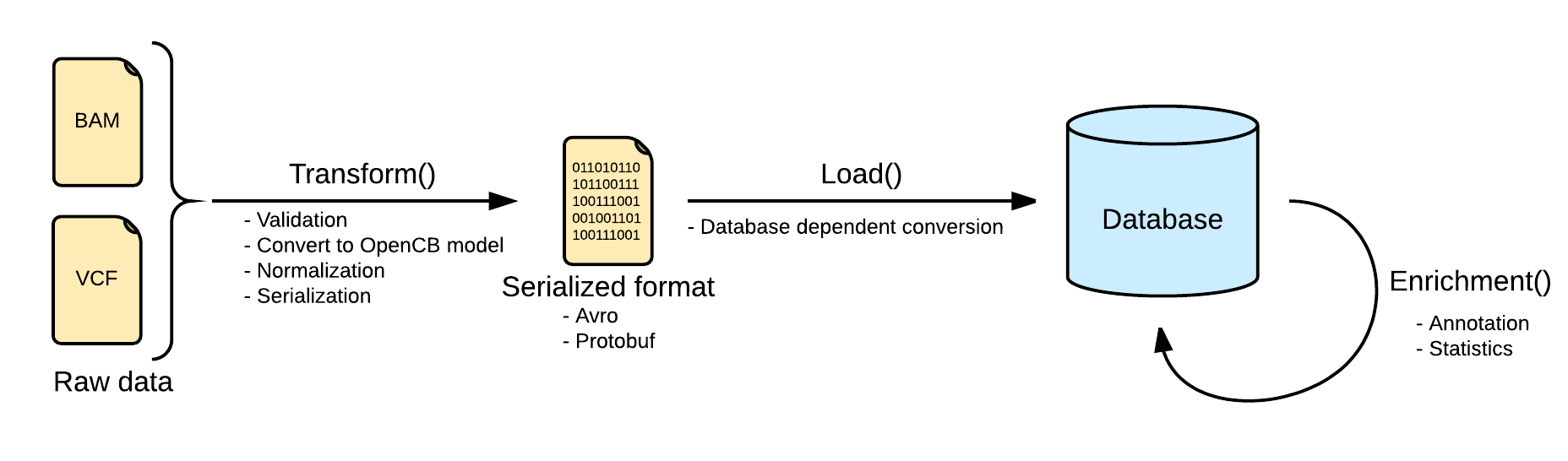
Transform
Files are converted Biodata models. The metadata and the data are serialized into two separated files. The metadata is stored into a file named <inputFileName>.file.json.gz serializing in json a single instance of the biodata model VariantSource, which mainly contains the header and some general stats. Along with this file, the real variants data is stored in a file named <inputFileName>.variants.avro.gz with a set of variant records described as the biodata model Variant.
VCF files are read using the library HTSJDK, which provides a syntactic validation of the data. Further actions on the validation will be taken, like duplicate or overlapping variants detection.
By default, malformed variants will be skipped and written into a third optional file named <inputFileName>.malformed.txt . If the transform step generates this file, a curation process should be taken to repair the file. Otherwise, the variants would be skipped.
All the variants in the transform step will be normalized as defined here: Variant Normalization. This will help to unify the variants representation, since the VCF specification allows multiple ways of referring to a variant and some ambiguities.
Load
Loading variants from multiple files into a single database will effectively merge them. In most of the scenarios, with a good normalization, merging variants is strait forward. But in some other scenarios, with multiple alternates or overlapping variants, the merge requires more logic. More information at Merge Mode.
Details about load are dependent on the implementation.
Limitations
- You can not load two files with the same sample in the same study. See OpenCGA#158.
There is an exception for this limitation for the scenarios where the variants were split in multiple files (by chromosome, by type, ...). In this case, you can use the parameter--load-split-data. SeeOpenCGA#696 - You can not index two files with the same name (e.g. /data/sample1/my.vcf.gz and /data/sample2/my.vcf.gz) in the same study. This limitation should not be a problem in any real scenario, where every VCF file usually has a different name. If two files have the same name, the most likely situation is that they contain the same samples, and this is already forbidden by the previous limitation.
Annotate
As part of the enrichment step, some extra information can be added to the variants database as Annotations. This VariantAnnotation can be fetch from Cellbase or read from local file provided by the user. The model of the variant annotation is defined in the project Biodata, in variantAnnotation.avdl
Annotators
Variant Storage Engine can make use of different annotators to produce the annotation for the variants.
The annotator can be modified at the annotating step, and the default value is defined in the storage-configuration.yml file:
- annotator: "cellbase_rest"
WARN Previous to version v1.3.0: Parameter "annotationSource" should be used instead of "annotator". See OpenCGA#747.
CellBase Annotator
PENDING
CellBase REST Annotator
This is the default annotator for OpenCGA. This Annotator connects to a CellBase installation using the REST API.
This is an example of cellbase annotation using a REST call:
CellBase Direct Annotator
The CellBaseDirectAnnotator creates a connection directly with the CellBase database. This requires a local installation of CellBase, which takes some resources, but it speeds up the annotation step removing network time.
Configuration
PENDING
- annotator.cellbase.exclude: "expression,hgvs,repeats,cytoband"
- annotator.cellbase.use_cache: true
- annotator.cellbase.imprecise_variants: false # Imprecise variants supported by cellbase (REST only)
Custom annotator
Custom annotation
The VariantAnnotation model includes a field for adding extra annotation attributes. This field is intended to contain custom annotation provided by the end user.
Additional attributes can be grouped by source. Each source will contain a set of key-value attributes creating this structure:
VariantAnnotation = {
// ...
"additionalAttributes" : {
"<source1>" : {
"attribute" : {
"<key1>":"<value>",
"<key2>":"<value>",
"<key3>":"<value>"
}
},
"<source2>" : {
"attribute" : {
"<key1>":"<value>",
"<key2>":"<value>",
"<key3>":"<value>"
}
}
}
OpenCGA Storage is able to load this custom annotation from 3 different formats: GFF, BED and VCF. When loading the new annotation data, the user has to provide a name for the new custom annotation. Because the structure of these file formats is slightly different, the information loaded won't be the same.
GFF and BED files describe features within a region, providing a chromosome, start and end. All the variants between the start and end will be annotated with the information.
GFF : From this file format, only the third column, containing the feature is extracted and loaded with the key "feature"
This line of GFF will generate the next additionalAttributes:GFFchr22 TeleGene enhancer 16053659 16063659 500 + . touch1
ResultVariantAnnotation = { // ... "additionalAttributes" : { "myGff" : { "attribute" : { "feature" : "enhancer" } } } }
BED : From the bed format, columns name (4th), score (5th) and strand (6th) will be loaded.
This line of BED will generate the next additionalAttributes:
BEDchr22 16053659 16063659 Pos1 353 + 127471196 127472363 255,0,0 0 A A
ResultVariantAnnotation = { // ... "additionalAttributes" : { "myBed" : { "attribute" : { "name":"Pos1", "score":"353", "strand":"+" } } } }
VCF : This format is not region based, so each line will modify a single variant. All the INFO column will be loaded as additional attributes.
The next VCF will generate the next additionalAttributes:
VCF##fileformat=VCFv4.2 ##FILTER=<ID=PASS,Description="All filters passed"> ##INFO=<ID=FEATURE,Number=1,Type=String,Description="Feature type"> ##INFO=<ID=SCORE,Number=1,Type=Integer,Description="Score value"> ##INFO=<ID=STRAND,Number=1,Type=Integer,Description="Strand"> #CHROM POS ID REF ALT QUAL FILTER INFO chr22 16050075 A G . 100 PASS FEATURE=specific;SCORE=300;STRAND=+
ResultVariantAnnotation = { // ... "additionalAttributes" : { "myVcf" : { "attribute" : { "FEATURE":"specific", "SCORE":"300", "STRAND":"+" } } } }
- Example with multiple sources: In case of having custom annotations from more than one source, more than one source will appear in the additionalAttributes field:Result
VariantAnnotation = { // ... "additionalAttributes" : { "myVcf" : { "attribute" : { "FEATURE":"specific", "SCORE":"300", "STRAND":"+" } }, "myBed" : { "attribute" : { "name":"Pos1", "score":"353", "strand":"+" } } } }
Calculate Statistics
Pre-calculated stats are useful for filtering variants. This stats are intra-study, calculated within a given cohort.
Cohorts
Cohorts are defined as a arbitrary group of samples. Cohorts can be defined in Catalog, either selecting samples one by one or selecting all samples that share some attributes like population or phenotype.
If a cohort is modified after calculating the statistics, the existing statistics became INVALID.
By default, in each study, there is defined the cohort ALL that contains all the samples loaded in the study. Every time that new samples are loaded in the study, this cohort is modified, and the statistics have to be recomputed.
Stats models
There are two types of statistics, per variant, and global statistics. Variant statistics are stored in the variants database, within the StudyEntry. Global statistics are stored in Catalog.
Variant Stats (intra variant)
These stats are calculated for each variant, and for a set of samples (cohort).ResultVariantStats // Total number of alleles in called genotypes. Do not include missing alleles int alleleCount // Number of reference alleles found in this variants int refAlleleCount // Number of main alternate alleles found in this variants. Do not include secondary alternates int altAlleleCount // Reference allele frequency calculated from refAlleleCount and alleleCount, in the range (0,1) float refAlleleFreq // Alternate allele frequency calculated from altAlleleCount and alleleCount, in the range (0,1) float altAlleleFreq // Count for each genotype found map<int> genotypeCount // Genotype frequency for each genotype found map<float> genotypeFreq // Number of missing alleles int missingAlleleCount // Number of missing genotypes int missingGenotypeCount // Minor allele frequency float maf // Minor genotype frequency float mgf // Allele with minor frequency string mafAllele // Genotype with minor frequency string mgfGenotype
Aggregated statistics
Usually, public studies do not provide samples data. In this situations is not possible to calculate the statistics. Instead, the statistics can be extracted from the INFO column. Unfortunately, there is no standard way for defining multi-cohort statistics in the VCF format. Instead, OpenCGA recognizes three different formats for representing statistics.
- BASIC mode
- EVS mode
- EXAC mode
Remove
Export / Query and variant filter
The main goal for indexing variant data into OpenCGA Storage is to be able to make queries and extract this data in a efficient way. This operation, executed via gRPC or with direct connection, allows to export a large quantity of variants from a database. It can work together with Import, be used only to provide input data to external analysis, or generate reports.
See Querying Variant Data to see all the possible filters over variants.
When exporting variants, some metadata files are generated, containing information regarding the studies, files and samples from the exported data.
There are multiple possible output formats:
- VCF
- JSON
- AVRO
Export frequencies (statistics)
Export frequencies (statistics) is an special case of export. Instead of export full variants, only the variant cohort statistics are exported.
To export variant frequencies, use the command variant export-frequencies in the command line.
opencga-analysis.sh variant export-frequencies -s <study> --output-format <vcf|tsv|cellbase|json> opencga-storage.sh variant export-frequencies -s <study> --output-format <vcf|tsv|cellbase|json
As for variants export, there are multiple possible output formats:
VCF : Standard VCF format without samples information, with the stats as values in the INFO column.
VCF##fileformat=VCFv4.2 ##FILTER=<ID=.,Description="No FILTER info"> ##FILTER=<ID=PASS,Description="Valid variant"> ##INFO=<ID=AC,Number=A,Type=Integer,Description="Total number of alternate alleles in called genotypes, for each ALT allele, in the same order as listed"> ##INFO=<ID=AF,Number=A,Type=Float,Description="Allele Frequency, for each ALT allele, calculated from AC and AN, in the range (0,1), in the same order as listed"> ##INFO=<ID=AN,Number=1,Type=Integer,Description="Total number of alleles in called genotypes"> ##INFO=<ID=AFK_AF,Number=A,Type=Float,Description="Allele frequency in the C1 cohort calculated from AC and AN, in the range (0,1), in the same order as listed"> #CHROM POS ID REF ALT QUAL FILTER INFO 22 16050115 . G A . PASS AC=1;AF=0.001;AN=1000;AFK_AF=0.002008 22 16050213 . C T . PASS AC=1;AF=0.001;AN=1000;AFK_AF=0 22 16050319 . C T . PASS AC=1;AF=0.001;AN=1000;AFK_AF=0 22 16050607 . G A . PASS AC=2;AF=0.002;AN=1000;AFK_AF=0.004016
TSV (Tab Separated Values). Simple format with each cohort in one column.
TSV#CHR POS REF ALT ALL_AN ALL_AC ALL_AF ALL_HET ALL_HOM 22 16050213 C T 1000 1 0.001 0.002 0.0 22 16050607 G A 1000 2 0.002 0.004 0.0 22 16050740 A - 1000 1 0.001 0.002 0.0 22 16050840 C G 1000 13 0.013 0.026 0.0 22 16051075 G A 1000 2 0.002 0.004 0.0 22 16051249 T C 1000 91 0.091 0.162 0.01 22 16051453 A C 998 74 0.074 0.144 0.004 22 16051453 A G 926 2 0.002 0.144 0.004 22 16051723 A - 1000 12 0.012 0.024 0.0 22 16051816 T G 1000 2 0.002 0.004 0.0
JSON. Variant model just with minimal information and statistics.
JSON{"reference":"G","names":[],"chromosome":"22","alternate":"A","start":16050115,"annotation":null,"id":"22:16050115:G:A","type":"SNV","studies":[{"format":[],"samplesData":[],"studyId":"user@p1:s1","stats":{"C3":{"refAllele":"G","altAllele":"A","refAlleleCount":2,"altAlleleCount":0,"genotypesCount":{"0/0":1},"genotypesFreq":{"0/0":1.0},"missingAlleles":0,"missingGenotypes":0,"refAlleleFreq":1.0,"altAlleleFreq":0.0,"maf":0.0,"mgf":0.0,"mafAllele":"A","mgfGenotype":"1/1","passedFilters":false,"mendelianErrors":-1,"casesPercentDominant":-1.0,"controlsPercentDominant":-1.0,"casesPercentRecessive":-1.0,"controlsPercentRecessive":-1.0,"quality":-1.0,"numSamples":-1,"variantType":"SNV"},"ALL":{"refAllele":"G","altAllele":"A","refAlleleCount":999,"altAlleleCount":1,"genotypesCount":{"0/0":499,"0|1":1},"genotypesFreq":{"0/0":0.998,"0|1":0.002},"missingAlleles":0,"missingGenotypes":0,"refAlleleFreq":0.999,"altAlleleFreq":0.001,"maf":0.001,"mgf":0.0,"mafAllele":"A","mgfGenotype":"1/1","passedFilters":false,"mendelianErrors":-1,"casesPercentDominant":-1.0,"controlsPercentDominant":-1.0,"casesPercentRecessive":-1.0,"controlsPercentRecessive":-1.0,"quality":-1.0,"numSamples":-1,"variantType":"SNV"},"C4":{"refAllele":"G","altAllele":"A","refAlleleCount":-1,"altAlleleCount":-1,"genotypesCount":{},"genotypesFreq":{},"missingAlleles":0,"missingGenotypes":0,"refAlleleFreq":2.0,"altAlleleFreq":-1.0,"maf":-1.0,"mgf":-1.0,"mafAllele":null,"mgfGenotype":null,"passedFilters":false,"mendelianErrors":-1,"casesPercentDominant":-1.0,"controlsPercentDominant":-1.0,"casesPercentRecessive":-1.0,"controlsPercentRecessive":-1.0,"quality":-1.0,"numSamples":-1,"variantType":"SNV"},"C1":{"refAllele":"G","altAllele":"A","refAlleleCount":500,"altAlleleCount":0,"genotypesCount":{"0/0":250},"genotypesFreq":{"0/0":1.0},"missingAlleles":0,"missingGenotypes":0,"refAlleleFreq":1.0,"altAlleleFreq":0.0,"maf":0.0,"mgf":0.0,"mafAllele":"A","mgfGenotype":"1/1","passedFilters":false,"mendelianErrors":-1,"casesPercentDominant":-1.0,"controlsPercentDominant":-1.0,"casesPercentRecessive":-1.0,"controlsPercentRecessive":-1.0,"quality":-1.0,"numSamples":-1,"variantType":"SNV"},"C2":{"refAllele":"G","altAllele":"A","refAlleleCount":497,"altAlleleCount":1,"genotypesCount":{"0/0":248,"0|1":1},"genotypesFreq":{"0/0":0.99598396,"0|1":0.004016064},"missingAlleles":0,"missingGenotypes":0,"refAlleleFreq":0.997992,"altAlleleFreq":0.002008032,"maf":0.002008032,"mgf":0.0,"mafAllele":"A","mgfGenotype":"1/1","passedFilters":false,"mendelianErrors":-1,"casesPercentDominant":-1.0,"controlsPercentDominant":-1.0,"casesPercentRecessive":-1.0,"controlsPercentRecessive":-1.0,"quality":-1.0,"numSamples":-1,"variantType":"SNV"}},"files":[],"secondaryAlternates":[]}],"end":16050115,"strand":"+","sv":null,"hgvs":{},"length":1} {"reference":"C","names":[],"chromosome":"22","alternate":"T","start":16050213,"annotation":null,"id":"22:16050213:C:T","type":"SNV","studies":[{"format":[],"samplesData":[],"studyId":"user@p1:s1","stats":{"C3":{"refAllele":"C","altAllele":"T","refAlleleCount":2,"altAlleleCount":0,"genotypesCount":{"0/0":1},"genotypesFreq":{"0/0":1.0},"missingAlleles":0,"missingGenotypes":0,"refAlleleFreq":1.0,"altAlleleFreq":0.0,"maf":0.0,"mgf":0.0,"mafAllele":"T","mgfGenotype":"1/1","passedFilters":false,"mendelianErrors":-1,"casesPercentDominant":-1.0,"controlsPercentDominant":-1.0,"casesPercentRecessive":-1.0,"controlsPercentRecessive":-1.0,"quality":-1.0,"numSamples":-1,"variantType":"SNV"},"ALL":{"refAllele":"C","altAllele":"T","refAlleleCount":999,"altAlleleCount":1,"genotypesCount":{"0|1":1,"0/0":499},"genotypesFreq":{"0|1":0.002,"0/0":0.998},"missingAlleles":0,"missingGenotypes":0,"refAlleleFreq":0.999,"altAlleleFreq":0.001,"maf":0.001,"mgf":0.0,"mafAllele":"T","mgfGenotype":"1/1","passedFilters":false,"mendelianErrors":-1,"casesPercentDominant":-1.0,"controlsPercentDominant":-1.0,"casesPercentRecessive":-1.0,"controlsPercentRecessive":-1.0,"quality":-1.0,"numSamples":-1,"variantType":"SNV"},"C4":{"refAllele":"C","altAllele":"T","refAlleleCount":-1,"altAlleleCount":-1,"genotypesCount":{},"genotypesFreq":{},"missingAlleles":0,"missingGenotypes":0,"refAlleleFreq":2.0,"altAlleleFreq":-1.0,"maf":-1.0,"mgf":-1.0,"mafAllele":null,"mgfGenotype":null,"passedFilters":false,"mendelianErrors":-1,"casesPercentDominant":-1.0,"controlsPercentDominant":-1.0,"casesPercentRecessive":-1.0,"controlsPercentRecessive":-1.0,"quality":-1.0,"numSamples":-1,"variantType":"SNV"},"C1":{"refAllele":"C","altAllele":"T","refAlleleCount":500,"altAlleleCount":0,"genotypesCount":{"0/0":250},"genotypesFreq":{"0/0":1.0},"missingAlleles":0,"missingGenotypes":0,"refAlleleFreq":1.0,"altAlleleFreq":0.0,"maf":0.0,"mgf":0.0,"mafAllele":"T","mgfGenotype":"1/1","passedFilters":false,"mendelianErrors":-1,"casesPercentDominant":-1.0,"controlsPercentDominant":-1.0,"casesPercentRecessive":-1.0,"controlsPercentRecessive":-1.0,"quality":-1.0,"numSamples":-1,"variantType":"SNV"},"C2":{"refAllele":"C","altAllele":"T","refAlleleCount":497,"altAlleleCount":1,"genotypesCount":{"0|1":1,"0/0":248},"genotypesFreq":{"0|1":0.004016064,"0/0":0.99598396},"missingAlleles":0,"missingGenotypes":0,"refAlleleFreq":0.997992,"altAlleleFreq":0.002008032,"maf":0.002008032,"mgf":0.0,"mafAllele":"T","mgfGenotype":"1/1","passedFilters":false,"mendelianErrors":-1,"casesPercentDominant":-1.0,"controlsPercentDominant":-1.0,"casesPercentRecessive":-1.0,"controlsPercentRecessive":-1.0,"quality":-1.0,"numSamples":-1,"variantType":"SNV"}},"files":[],"secondaryAlternates":[]}],"end":16050213,"strand":"+","sv":null,"hgvs":{},"length":1} {"reference":"C","names":[],"chromosome":"22","alternate":"T","start":16050319,"annotation":null,"id":"22:16050319:C:T","type":"SNV","studies":[{"format":[],"samplesData":[],"studyId":"user@p1:s1","stats":{"C3":{"refAllele":"C","altAllele":"T","refAlleleCount":2,"altAlleleCount":0,"genotypesCount":{"0/0":1},"genotypesFreq":{"0/0":1.0},"missingAlleles":0,"missingGenotypes":0,"refAlleleFreq":1.0,"altAlleleFreq":0.0,"maf":0.0,"mgf":0.0,"mafAllele":"T","mgfGenotype":"1/1","passedFilters":false,"mendelianErrors":-1,"casesPercentDominant":-1.0,"controlsPercentDominant":-1.0,"casesPercentRecessive":-1.0,"controlsPercentRecessive":-1.0,"quality":-1.0,"numSamples":-1,"variantType":"SNV"},"ALL":{"refAllele":"C","altAllele":"T","refAlleleCount":999,"altAlleleCount":1,"genotypesCount":{"0/0":499,"1|0":1},"genotypesFreq":{"0/0":0.998,"1|0":0.002},"missingAlleles":0,"missingGenotypes":0,"refAlleleFreq":0.999,"altAlleleFreq":0.001,"maf":0.001,"mgf":0.0,"mafAllele":"T","mgfGenotype":"1/1","passedFilters":false,"mendelianErrors":-1,"casesPercentDominant":-1.0,"controlsPercentDominant":-1.0,"casesPercentRecessive":-1.0,"controlsPercentRecessive":-1.0,"quality":-1.0,"numSamples":-1,"variantType":"SNV"},"C4":{"refAllele":"C","altAllele":"T","refAlleleCount":-1,"altAlleleCount":-1,"genotypesCount":{},"genotypesFreq":{},"missingAlleles":0,"missingGenotypes":0,"refAlleleFreq":2.0,"altAlleleFreq":-1.0,"maf":-1.0,"mgf":-1.0,"mafAllele":null,"mgfGenotype":null,"passedFilters":false,"mendelianErrors":-1,"casesPercentDominant":-1.0,"controlsPercentDominant":-1.0,"casesPercentRecessive":-1.0,"controlsPercentRecessive":-1.0,"quality":-1.0,"numSamples":-1,"variantType":"SNV"},"C1":{"refAllele":"C","altAllele":"T","refAlleleCount":500,"altAlleleCount":0,"genotypesCount":{"0/0":250},"genotypesFreq":{"0/0":1.0},"missingAlleles":0,"missingGenotypes":0,"refAlleleFreq":1.0,"altAlleleFreq":0.0,"maf":0.0,"mgf":0.0,"mafAllele":"T","mgfGenotype":"1/1","passedFilters":false,"mendelianErrors":-1,"casesPercentDominant":-1.0,"controlsPercentDominant":-1.0,"casesPercentRecessive":-1.0,"controlsPercentRecessive":-1.0,"quality":-1.0,"numSamples":-1,"variantType":"SNV"},"C2":{"refAllele":"C","altAllele":"T","refAlleleCount":497,"altAlleleCount":1,"genotypesCount":{"0/0":248,"1|0":1},"genotypesFreq":{"0/0":0.99598396,"1|0":0.004016064},"missingAlleles":0,"missingGenotypes":0,"refAlleleFreq":0.997992,"altAlleleFreq":0.002008032,"maf":0.002008032,"mgf":0.0,"mafAllele":"T","mgfGenotype":"1/1","passedFilters":false,"mendelianErrors":-1,"casesPercentDominant":-1.0,"controlsPercentDominant":-1.0,"casesPercentRecessive":-1.0,"controlsPercentRecessive":-1.0,"quality":-1.0,"numSamples":-1,"variantType":"SNV"}},"files":[],"secondaryAlternates":[]}],"end":16050319,"strand":"+","sv":null,"hgvs":{},"length":1} {"reference":"G","names":[],"chromosome":"22","alternate":"A","start":16050607,"annotation":null,"id":"22:16050607:G:A","type":"SNV","studies":[{"format":[],"samplesData":[],"studyId":"user@p1:s1","stats":{"C3":{"refAllele":"G","altAllele":"A","refAlleleCount":2,"altAlleleCount":0,"genotypesCount":{"0/0":1},"genotypesFreq":{"0/0":1.0},"missingAlleles":0,"missingGenotypes":0,"refAlleleFreq":1.0,"altAlleleFreq":0.0,"maf":0.0,"mgf":0.0,"mafAllele":"A","mgfGenotype":"1/1","passedFilters":false,"mendelianErrors":-1,"casesPercentDominant":-1.0,"controlsPercentDominant":-1.0,"casesPercentRecessive":-1.0,"controlsPercentRecessive":-1.0,"quality":-1.0,"numSamples":-1,"variantType":"SNV"},"ALL":{"refAllele":"G","altAllele":"A","refAlleleCount":998,"altAlleleCount":2,"genotypesCount":{"0/0":498,"0|1":1,"1|0":1},"genotypesFreq":{"0/0":0.996,"0|1":0.002,"1|0":0.002},"missingAlleles":0,"missingGenotypes":0,"refAlleleFreq":0.998,"altAlleleFreq":0.002,"maf":0.002,"mgf":0.0,"mafAllele":"A","mgfGenotype":"1/1","passedFilters":false,"mendelianErrors":-1,"casesPercentDominant":-1.0,"controlsPercentDominant":-1.0,"casesPercentRecessive":-1.0,"controlsPercentRecessive":-1.0,"quality":-1.0,"numSamples":-1,"variantType":"SNV"},"C4":{"refAllele":"G","altAllele":"A","refAlleleCount":-1,"altAlleleCount":-1,"genotypesCount":{},"genotypesFreq":{},"missingAlleles":0,"missingGenotypes":0,"refAlleleFreq":2.0,"altAlleleFreq":-1.0,"maf":-1.0,"mgf":-1.0,"mafAllele":null,"mgfGenotype":null,"passedFilters":false,"mendelianErrors":-1,"casesPercentDominant":-1.0,"controlsPercentDominant":-1.0,"casesPercentRecessive":-1.0,"controlsPercentRecessive":-1.0,"quality":-1.0,"numSamples":-1,"variantType":"SNV"},"C1":{"refAllele":"G","altAllele":"A","refAlleleCount":499,"altAlleleCount":1,"genotypesCount":{"0/0":249,"0|1":1},"genotypesFreq":{"0/0":0.996,"0|1":0.004},"missingAlleles":0,"missingGenotypes":0,"refAlleleFreq":0.998,"altAlleleFreq":0.002,"maf":0.002,"mgf":0.0,"mafAllele":"A","mgfGenotype":"1/1","passedFilters":false,"mendelianErrors":-1,"casesPercentDominant":-1.0,"controlsPercentDominant":-1.0,"casesPercentRecessive":-1.0,"controlsPercentRecessive":-1.0,"quality":-1.0,"numSamples":-1,"variantType":"SNV"},"C2":{"refAllele":"G","altAllele":"A","refAlleleCount":497,"altAlleleCount":1,"genotypesCount":{"0/0":248,"1|0":1},"genotypesFreq":{"0/0":0.99598396,"1|0":0.004016064},"missingAlleles":0,"missingGenotypes":0,"refAlleleFreq":0.997992,"altAlleleFreq":0.002008032,"maf":0.002008032,"mgf":0.0,"mafAllele":"A","mgfGenotype":"1/1","passedFilters":false,"mendelianErrors":-1,"casesPercentDominant":-1.0,"controlsPercentDominant":-1.0,"casesPercentRecessive":-1.0,"controlsPercentRecessive":-1.0,"quality":-1.0,"numSamples":-1,"variantType":"SNV"}},"files":[],"secondaryAlternates":[]}],"end":16050607,"strand":"+","sv":null,"hgvs":{},"length":1}Population Frequencies (Cellbase mode). Specific JSON format for import into Cellbase variation. It is a Variant model with VariantAnnotation with PupulationFrequencies.
PopulationFrequencies / Cellbase{"names":[],"reference":"T","chromosome":"22","alternate":"C","start":16174643,"annotation":{"populationFrequencies":[{"study":"s1","population":"ALL","refAllele":"T","altAllele":"C","refAlleleFreq":0.999,"altAlleleFreq":0.001,"refHomGenotypeFreq":0.998,"hetGenotypeFreq":0.002,"altHomGenotypeFreq":0.0},{"study":"s1","population":"C1","refAllele":"T","altAllele":"C","refAlleleFreq":0.998,"altAlleleFreq":0.002,"refHomGenotypeFreq":0.996,"hetGenotypeFreq":0.004,"altHomGenotypeFreq":0.0}]},"end":16174643,"type":"SNV","studies":[],"strand":"+","hgvs":{},"length":1} {"names":[],"reference":"C","chromosome":"22","alternate":"T","start":16176715,"annotation":{"populationFrequencies":[{"study":"s1","population":"ALL","refAllele":"C","altAllele":"T","refAlleleFreq":0.998,"altAlleleFreq":0.002,"refHomGenotypeFreq":0.996,"hetGenotypeFreq":0.004,"altHomGenotypeFreq":0.0},{"study":"s1","population":"C2","refAllele":"C","altAllele":"T","refAlleleFreq":0.99598396,"altAlleleFreq":0.004016064,"refHomGenotypeFreq":0.99196786,"hetGenotypeFreq":0.008032128,"altHomGenotypeFreq":0.0}]},"end":16176715,"type":"SNV","studies":[],"strand":"+","hgvs":{},"length":1} {"names":[],"reference":"C","chromosome":"22","alternate":"A","start":16176724,"annotation":{"populationFrequencies":[{"study":"s1","population":"ALL","refAllele":"C","altAllele":"A","refAlleleFreq":0.999,"altAlleleFreq":0.001,"refHomGenotypeFreq":0.998,"hetGenotypeFreq":0.002,"altHomGenotypeFreq":0.0},{"study":"s1","population":"C2","refAllele":"C","altAllele":"A","refAlleleFreq":0.997992,"altAlleleFreq":0.002008032,"refHomGenotypeFreq":0.99598396,"hetGenotypeFreq":0.004016064,"altHomGenotypeFreq":0.0}]},"end":16176724,"type":"SNV","studies":[],"strand":"+","hgvs":{},"length":1} {"names":[],"reference":"T","chromosome":"22","alternate":"C","start":16176769,"annotation":{"populationFrequencies":[{"study":"s1","population":"ALL","refAllele":"T","altAllele":"C","refAlleleFreq":0.999,"altAlleleFreq":0.001,"refHomGenotypeFreq":0.998,"hetGenotypeFreq":0.002,"altHomGenotypeFreq":0.0},{"study":"s1","population":"C2","refAllele":"T","altAllele":"C","refAlleleFreq":0.997992,"altAlleleFreq":0.002008032,"refHomGenotypeFreq":0.99598396,"hetGenotypeFreq":0.004016064,"altHomGenotypeFreq":0.0}]},"end":16176769,"type":"SNV","studies":[],"strand":"+","hgvs":{},"length":1} {"names":[],"reference":"T","chromosome":"22","alternate":"A","start":16176926,"annotation":{"populationFrequencies":[{"study":"s1","population":"C3","refAllele":"T","altAllele":"A","refAlleleFreq":0.5,"altAlleleFreq":0.5,"refHomGenotypeFreq":0.0,"hetGenotypeFreq":1.0,"altHomGenotypeFreq":0.0},{"study":"s1","population":"ALL","refAllele":"T","altAllele":"A","refAlleleFreq":0.473,"altAlleleFreq":0.527,"refHomGenotypeFreq":0.166,"hetGenotypeFreq":0.614,"altHomGenotypeFreq":0.22},{"study":"s1","population":"C1","refAllele":"T","altAllele":"A","refAlleleFreq":0.474,"altAlleleFreq":0.526,"refHomGenotypeFreq":0.164,"hetGenotypeFreq":0.62,"altHomGenotypeFreq":0.216},{"study":"s1","population":"C2","refAllele":"T","altAllele":"A","refAlleleFreq":0.4698795,"altAlleleFreq":0.5301205,"refHomGenotypeFreq":0.16465864,"hetGenotypeFreq":0.6104418,"altHomGenotypeFreq":0.2248996}]},"end":16176926,"type":"SNV","studies":[],"strand":"+","hgvs":{},"length":1}
Import
Table of Contents: