Here you can find a full report of about loading 62,000 samples for Genomics England Research environment.
Platform
The platform used for this case study consists on a Hadoop Cluster of 35 nodes (5 + 30) and a LSF queue system:
| Node | #nodes | cores | memory (GB) |
|---|---|---|---|
| LSF queue node for load | 10 | 12 | 364 |
| Hadoop master nodes | 5 | 28 | 216 |
| Hadoop worker nodes | 30 | 28 | 216 |
Data
The data of this case study contains a total of 64,078 samples divided in 4 different datasets.
| Dataset | Alias | Files | File type | Samples | Samples per file | Variants |
|---|---|---|---|---|---|---|
| Rare Disease GRCh37 | RD37 | 5,329 | Multi sample VCF | 12,142 | 2.28 | 298,763,059 |
| Rare Disease GRCh38 | RD38 | 16,591 | Multi sample VCF | 33,180 | 2.00 | 437,740,498 |
| Cancer Germline GRCh38 | CG38 | 9,167 | Single sample VCF | 9,167 | 1.00 | 286,136,051 |
| Cancer Somatic GRCh38 | CS38 | 9,589 | Somatic VCF | 9,589 | 1.00 | 398,402,166 |
| Total | 40,676 | 64,078 | 1,421,041,774 | |||
Each dataset is loaded in OpenCGA as a study.
Loading Data
The data ingestion is executed in the LSF nodes, connected directly to the Hadoop platform. This configuration allows us to run multiple files at the same time.
Having 10 worker nodes in the queue, each of them loading up to 6 files at the same time, results in 60 files being loaded concurrently.
Multi sample files
The files from Rare Disease studies (RD38 & RD37) contain more than one sample per file. In average, 2 samples per file.
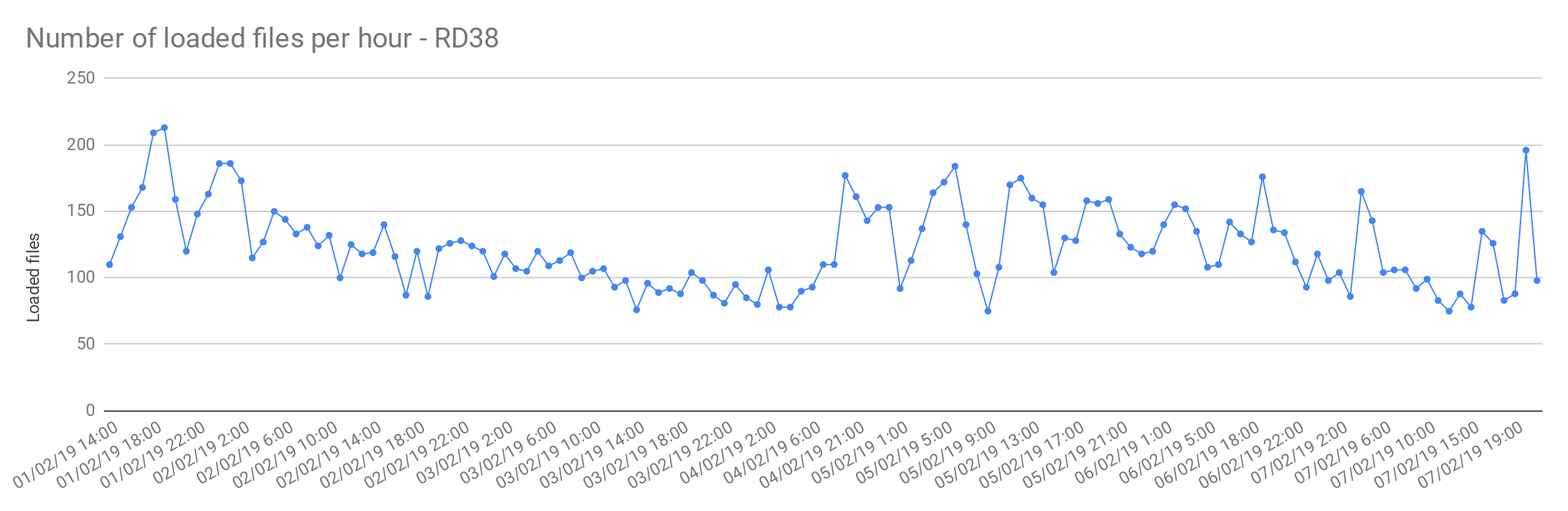
| Concurrent load files | 60 |
|---|---|
| Average files loaded per hour | 125.72 |
| Load time per file | 00:28:38 |
Single sample files
The files from Cancer Germline studies (CG38) contain one sample per file. Compared with the Rare Disease, these files are smaller in size, therefore, the load is slightly faster.
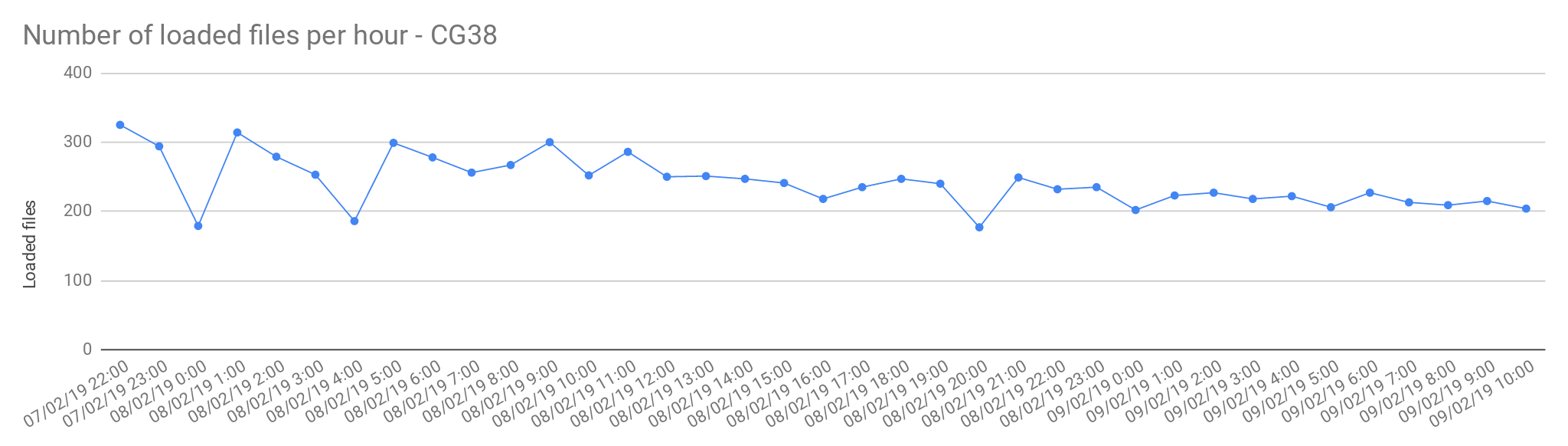
| Concurrent load files | 60 |
|---|---|
| Average files loaded per hour | 242.05 |
| Load time per file | 00:14:52 |
Query Performance
Common Queries
Clinical Queries
Table of Contents: