Overview
Index pipeline is the process of ingesting data into an OpenCGA-Storage backend. We define a general pipeline that is used and extended for the supported bioformats like variants and alignments. This pipeline can be extended by additional steps of enrichment, which will be highly dependent on the file format. At the end, the data may be filtered to be visualized, or used as analysis input data.
This concept is represented in Catalog to help the tracking of this status in different files.
Index
Indexing data pipeline consists in three steps, transforming the input raw data into an intermediate format, loading it into the selected database, depending on the implementation, and adding more information to the loaded data by calculating statistics or adding extra information like annotation.
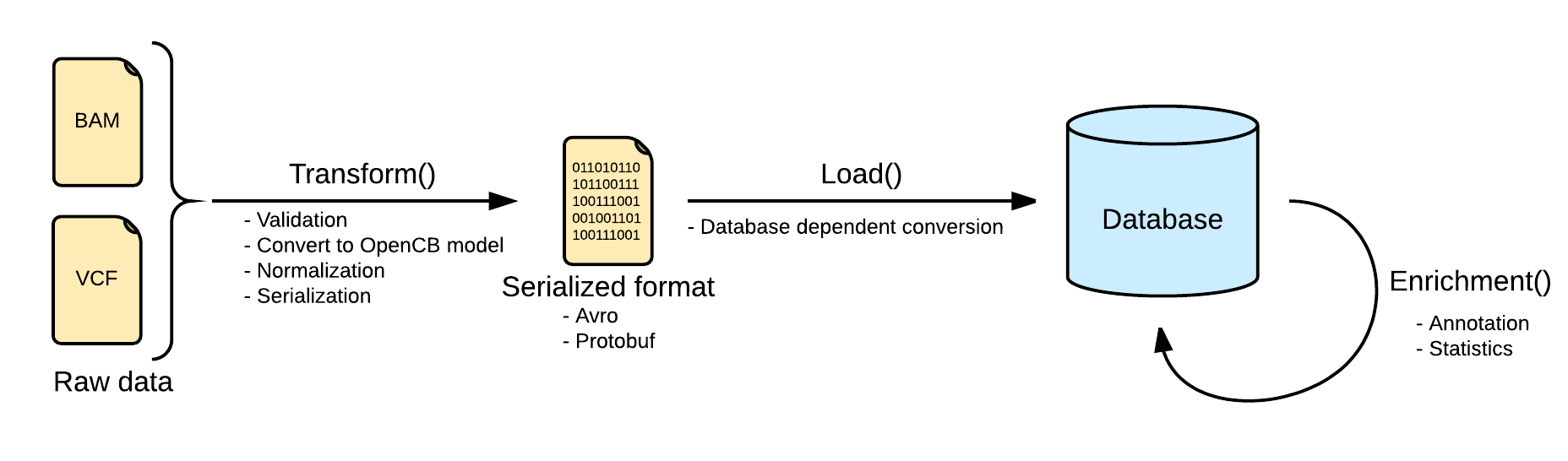
Transform
The first and one of the most important steps is the transformation. At this point, the pipeline ensures that the input file is valid and can be loaded into the database. The input file is read and converted into the OpenCB models, defined in Biodata. One single input file may generate more than one file, separating the data from the metadata. See Data Models for more info.
Depending on the input data the process will be more or less complex, and, at the end, the file will be serialized into disk using some serialization schema like Avro, Protobuf or Json in some cases.
As the transformation stage grants that the data is valid and can be loaded, the load stage can not start until the transformation has finished.
This step is shared between all the storage engine implementations of the same bioformat, so the result should be valid for any implementation.
Load
This step is intended to be as fast as possible, to avoid unnecessary downtimes in the database due to the work load. Because of this, all the convert and validate operations are made in the previous step.
Most of the storage engines are not going to load directly the opencb models, and some more engine dependent transformations are still expected. The storage engines grant that any valid instance of the input data model can be transformed and loaded.
In some scenarios the load step may be done in two steps, loading first into an intermediate system a batch of files, and then, move all of them to the real storage system. This could improve the loading speed by consuming more storage resources.
Enrichment
Despite input file formats contains a lot of interesting information, some of them is not directly available, and has to be calculated or fetched from external services.
Most of the storage engines provides mechanisms to calculate some statistics (either per record or aggregated) and store them back in the database to help the filtering process. By doing this, we can speed up some queries against pre-calculated statistics.
Some other information can not be inferred from the input data, and has to be feched from external annotation services like Cellbase.
Variant index pipeline
Indexing variants does not apply any modification to this generic pipeline. The input file format is VCF, accepting different variations like gVCF or aggregated VCFs.
Index
Transform
Files are converted Biodata models. The metadata and the data are serialized into two separated files. The metadata is stored into a file named <inputFileName>.file.json.gzserializing in json a single instance of the biodata model VariantSource, which mainly contains the header and some general stats. Along with this file, the real variants data is stored in a file named <inputFileName>.variants.avro.gz with a set of variant records described as the biodata model Variant.
VCF files are read using the library HTSJDK, which provides a syntactic validation of the data. Further actions on the validation will be taken, like duplicate or overlapping variants detection.
By default, malformed variants will be skipped and written into a third optional file named <inputFileName>.malformed.txt . If the transform step generates this file, a curation process should be taken to repair the file. Otherwise, the variants would be skipped.
All the variants in the transform step will be normalized as defined here: Variant Normalization. This will help to unify the variants representation, since the VCF specification allows multiple ways of referring to a variant and some ambiguities.
Load
Loading variants from multiple files into a single database will effectively merge them. In most of the scenarios, with a good normalization, merging variants is strait forward. But in some other scenarios, with multiple alternates or overlapping variants, the merge requires more logic. More information at Variant Merging.
Details about load are dependent on the implementation.
Annotation
As part of the enrichment step, some extra information can be added to the variants database as Annotations. This VariantAnnotation can be fetch from Cellbase or read from local file provided by the user.
More information at Variant Annotation and Stats.
Stats calculation
Pre-calculated stats are useful for filtering variants.
More information at Variant Annotation and Stats.