Genomic and Clinical Data
For this proof of concept (PoC) we loaded all the genomic variants of about 4,700 genomes from Genomics England, variants were loaded and indexed in the development version OpenCGA 2.0.0-beta. In total we loaded about 208 million unique variants from 4,700 gVCF files accounting for about 20TB of compressed disk space. It is worth noting that these files were generated using Dragen 2.x and the are unusually big, about 5-6GB per file.
Platform
For this proof of concept (PoC) we used the development version OpenCGA v2.0.0-beta using the Hadoop Variant Storage Engine that uses Apache HBase as back-end. We also used CellBase 4.6 for the variant annotation.
For the platform we used a 10-nodes Azure HDInsight 3.6 cluster using Data Lake Storage Gen2. HDInsight 3.6 uses Hortonworks HDP 2.6.5 (with Hadoop 2.7.3 and HBase 1.1.2) and we used Azure Batch for loading concurrently all the VCF files which had been copied previously to a NFS server, you can see details here:
| Node Type | Nodes | Azure Type | Cores | Memory (GB) | Storage |
|---|---|---|---|---|---|
| Hadoop Master | 3 | Standard_D12_V2 | 4 | 28 | Data Lake Gen2 |
| Hadoop Worker | 10 | Standard_DS13_V2 | 8 | 56 | Data Lake Gen2 |
| Azure Batch Queue | 20 | Standard_D4s_v3 | 4 | 16 | NFS Server |
Initially, we evaluated the new HDInsight 4.0, although it worked quite well there were some few minor issue, so for this PoC we decided to use the more stable HDInsight 3.6 (HDI3.6) over Data Lake Gen2 (DL2), we will refer to this as HDI3.6+DL2. During the PoC we worked with Azure engineers to debug and fix all these issues, unfortunately we did no have time to repeat the benchmark.
As you will see below in the Analysis Benchmark section, once we completed the PoC we increased the size of HDInsight to 20 nodes and repeated some tests to study the performance improvement.
Genomic Data Loading and Indexing
In order to study the loading performance we set up a Azure Batch queue of 20 computing nodes. This allowed us to load multiple files at the same time from different servers. We configured Azure Batch to load 1 VCF file per node resulting in 20 files being loaded in HBase simultaneously. During the load we studied two different configurations:
- Loading VCF files vs. loading transformed files
- Using HDD vs. SSD disk in the NFS server
You can observe the results in the following plot:
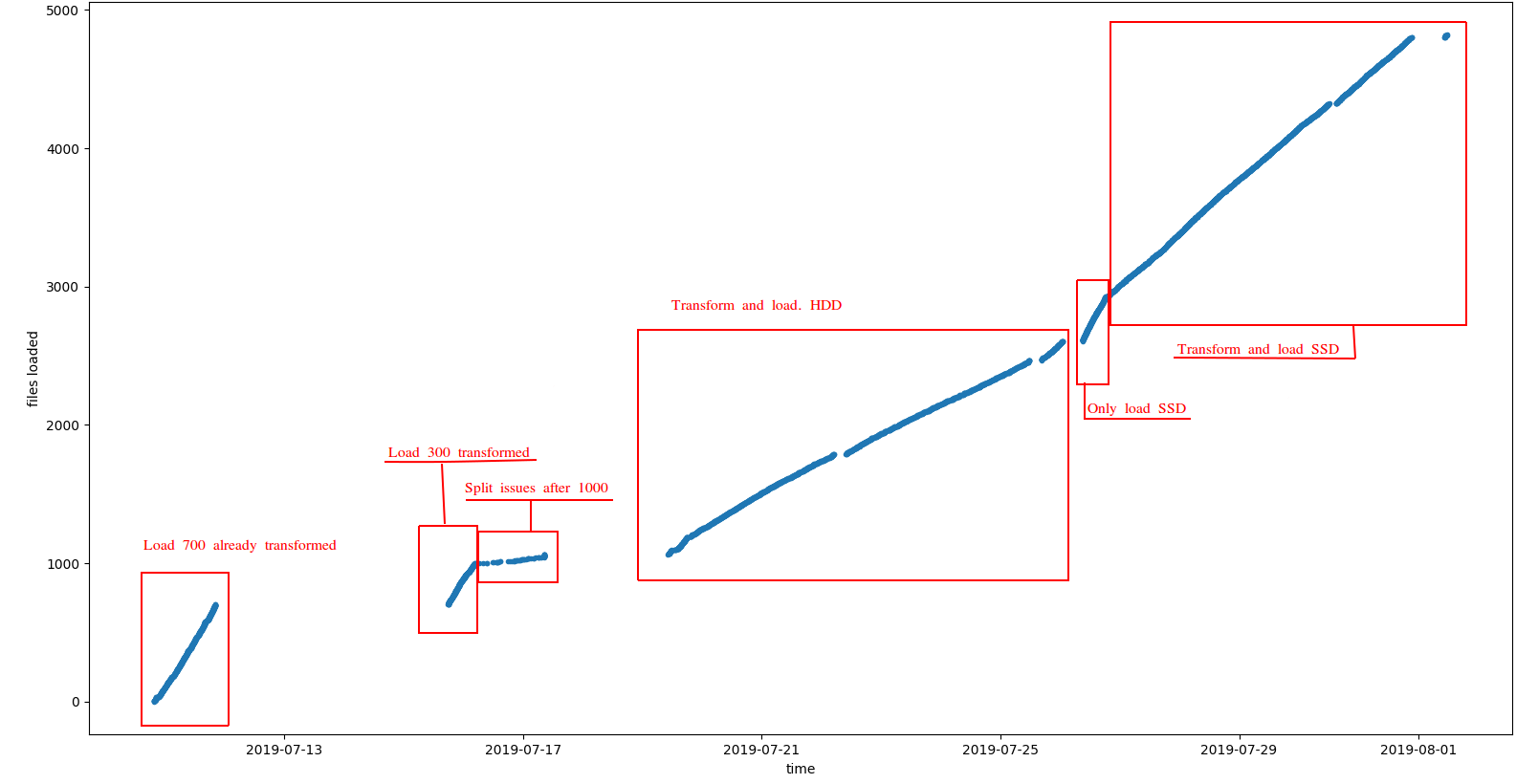
Some comments:
- As expected loading already transformed files is much faster since we only need to load and index data in HBase. See this link for more information Indexing Genomic Variants
- Also, loading from SSD disks showed a better performance
The most typical scenario when indexing genomic data is to transform+load at the same time, so assuming SSD disk the observed performance was about 380 VCF files indexed a day, or about 2TB/day. It is worth noting that:
- gVCF used were several times bigger than usual
- the number of Hadoop worker nodes was just 10
- we loaded up to 20 files concurrently but this could have been increased
These variables have a huge impact in the indexing performance, so the expected performance with more real gVCF files and production cluster is more than 1,000 VCF a day.
Analysis Benchmark
In this section you can find information about the performance of main variant storage engine operations, queries and analysis. Please, for data loading performance information go to section Genomic Data Load above.
Variant Storage Operations
Variant Storage operations take care of preparing the data for executing queries and analysis. Some of the most important operations include: Genotype Aggregation, Variant Annotation, Cohort Stats Calculation and Sample Variants Indexing.
Genotype Aggregation
We executed an initial aggregation with the first batch 700 samples accounting for 74.096.015 variants. This run in about 4:10 hours without any issue, this performance is expected to be quite stable. Also, the number of worker nodes affects the performance, unfortunately, because lack of time we could only test with 10 nodes.
Variant Annotation
This operation uses the CellBase to annotate each unique variant in the database, this annotation include consequence types, population frequencies, conservation scores clinical info, ... and will be typically used for variant queries and different analysis. We executed variant annotation during the variant indexing, in OpenCGA 2.0.0 you can load variants and annotate at the same time, the performance of variant annotation is affected mainly by CellBase installation, in this PoC we used a small CellBase installation from University of Cambridge which is far from ideal. The performance observed was about 120 million variants annotated a day.
Cohort Stats Calculation
We pre-computed the variants stats for the 200 million variants across the 4,700 samples, this operation includes the calculation and indexing of the variant stats. We run this few times with 10 nodes and 20 nodes. The observed performance was:
- With 10-nodes: 1:50 hours
- With 20-nodes: 53 min
As expected the performance improves linearly (2.07x speed-up) with the number of nodes.
Sample Variant Indexing
This operation plays a crucial role when querying by sample genotype. This is also one of the most complex and intensive operation since we index here all the genotypes loaded, all the variants for each sample is indexed, in this PoC we have about 20 billion variants across the 4,700 samples. We run this few times with 10 nodes and 20 nodes. The observed performance was:
- With 10-nodes: 4:53 hours
- With 20-nodes: 2:37 min
As expected the performance improves linearly (1.86x speed-up) with the number of nodes. Note: this operation is incremental so if we load new samples we only need to index variants from these samples, the runtime above was obtained indexing all sample genotypes in one single execution.
Query Benchmark
To study the query performance we tried different configurations:
- Variants table compression - Either GZ (2.9TB) or SNAPPY (4,7TB)
- Bucket cache size per node - Using the 1TB premium disk to improve writes to extend the bucket size
- Cache compression - HBASE-11331
- Cache warmup - Custom MR job to read the whole hbase table and warm up the caches
Queries
- filter=PASS,region,sample(1, OR)
- filter=PASS,region,sample(2, OR)
- filter=PASS,region,sample(3, OR)
- filter=PASS,region,sample(4, OR)
- filter=PASS,region,sample(5, OR)
- filter=PASS,region,sample(1, AND)
- filter=PASS,region,sample(2, AND)
- filter=PASS,region,sample(3, AND)
- filter=PASS,region,sample(4, AND)
- filter=PASS,region,sample(5, AND)
- filter=PASS,region=15,sample(3, AND)
- filter=PASS,region=15,sample(3, OR)
- filter=PASS,region,ct=(lof,missense_variant),sample(1, OR)
- filter=PASS,region,ct=(lof,missense_variant),sample(2, OR)
- filter=PASS,region,ct=(lof,missense_variant),sample(3, OR)
- filter=PASS,region,ct=(lof,missense_variant),sample(4, OR)
- filter=PASS,region,ct=(lof,missense_variant),sample(5, OR)
- filter=PASS,region,ct(6),sample(1, OR)
- filter=PASS,region,ct(6),sample(2, OR)
- filter=PASS,region,ct(6),sample(3, OR)
- filter=PASS,region,ct(6),sample(4, OR)
- filter=PASS,region,ct(6),sample(5, OR)
- filter=PASS,region,ct=(lof,missense_variant),biotype=protein_coding,sample(1, OR)
- filter=PASS,region,ct(9),biotype=protein_coding,sample(1, OR)
Scenarios
| ID | Variants Table Compression | Bucket Cache size (GB/node) | Cache compression | Cache warmup | Average Query Time (s) | Speed-up | |
|---|---|---|---|---|---|---|---|
| 1 | GZ | 45 | No | No | 5.487 | NA | |
| 2 | GZ | 45 | No | Yes | 5.126 | 1.1x | |
| 3 | GZ | 300 | No | Yes | 3.628 | 1.5x | |
| 4 | GZ | 300 | Yes | Yes | 2.115 | 2.6x | |
| 5 | SNAPPY | 300 | Yes | No | 2.412 | 2.3x | |
| 6 | SNAPPY | 300 | Yes | Yes | 1.816 | 3.0x | |
| 7 | SNAPPY | 45 | No | No | 3.750 | 1.5x |
Results
| AZURE | Native on-prem | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HDI3 | HDI4 | HDP2.6 | ||||||||||
| GZ | SNAPPY | SNAPPY | SNAPPY | |||||||||
| 45GB | 300GB | 300GB | 45GB | 500GB | 10GB | |||||||
| Uncompressed | Uncomp. | Comp. | Compressed | Comp. | Compressed | Uncomp. | ||||||
| Cold | Warm | Warm | Warm | Cold | Warm | Cold | Cold | Warm | RD37 | RD38 | CG38 | |
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | |
| filter=PASS,region,ct(6),sample(1, OR) | 5.172 | 5.729 | 4.263 | 1.792 | 2.950 | 1.534 | 3.561 | 4.446 | 1.495 | 0.829 | 2.729 | 1.246 |
| filter=PASS,region,ct(6),sample(2, OR) | 6.488 | 5.905 | 4.035 | 2.116 | 3.256 | 2.007 | 3.926 | 5.338 | 2.081 | 0.84 | 1.644 | 2.27 |
| filter=PASS,region,ct(6),sample(3, OR) | 7.657 | 5.366 | 4.383 | 2.715 | 3.299 | 2.725 | 4.355 | 6.427 | 2.285 | 1.947 | 2.205 | 1.99 |
| filter=PASS,region,ct(6),sample(4, OR) | 9.039 | 6.402 | 5.098 | 2.124 | 3.562 | 2.338 | 5.263 | 5.108 | 2.522 | 0.996 | 3.01 | 1.88 |
| filter=PASS,region,ct(6),sample(5, OR) | 8.688 | 8.546 | 4.880 | 3.161 | 3.432 | 2.455 | 5.778 | 7.006 | 3.067 | 1.628 | 1.961 | 1.962 |
| filter=PASS,region,ct(9),biotype=protein_coding,sample(1, OR) | 4.138 | 6.090 | 2.891 | 1.837 | 2.544 | 2.605 | 3.906 | NA* | NA* | 1.43 | 1.345 | 1.345 |
| filter=PASS,region,ct=(lof,missense_variant),biotype=protein_coding,sample(1, OR) | 3.908 | 5.060 | 3.284 | 1.795 | 1.804 | 1.588 | 3.619 | 3.758 | 1.714 | 0.802 | 1.549 | 1.443 |
| filter=PASS,region,ct=(lof,missense_variant),sample(1, OR) | 5.905 | 3.856 | 3.394 | 2.304 | 2.737 | 1.422 | 3.283 | 4.136 | 1.913 | 0.85 | 1.78 | 1.461 |
| filter=PASS,region,ct=(lof,missense_variant),sample(2, OR) | 7.762 | 5.686 | 3.996 | 2.561 | 3.348 | 2.017 | 3.912 | 4.209 | 2.375 | 1.765 | 2.533 | 4.981 |
| filter=PASS,region,ct=(lof,missense_variant),sample(3, OR) | 7.069 | 6.668 | 4.145 | 2.121 | 3.588 | 2.173 | 4.568 | 5.724 | 2.539 | 1.147 | 2.019 | 1.959 |
| filter=PASS,region,ct=(lof,missense_variant),sample(4, OR) | 6.918 | 6.488 | 4.908 | 3.000 | 3.867 | 2.622 | 5.013 | 5.987 | 3.065 | 1.229 | 2.01 | 1.754 |
| filter=PASS,region,ct=(lof,missense_variant),sample(5, OR) | 8.917 | 9.043 | 4.799 | 2.202 | 3.251 | 2.569 | 5.996 | 6.64 | 2.981 | 1.653 | 2.732 | 2.115 |
| filter=PASS,region,sample(1, AND) | 3.678 | 2.882 | 2.563 | 1.697 | 1.405 | 1.262 | 1.789 | 2.589 | 1.695 | 0.808 | 2.127 | 1.533 |
| filter=PASS,region,sample(1, OR) | 3.381 | 3.052 | 2.946 | 1.959 | 1.265 | 1.312 | 1.740 | 2.192 | 1.667 | 0.832 | 2.595 | 1.915 |
| filter=PASS,region,sample(2, AND) | 4.220 | 3.682 | 2.852 | 1.864 | 1.785 | 1.443 | 2.665 | 3.088 | 1.980 | 1.046 | 3.161 | 1.802 |
| filter=PASS,region,sample(2, OR) | 3.997 | 3.657 | 2.401 | 2.170 | 1.532 | 1.327 | 2.255 | 2.963 | 1.726 | 0.925 | 1.827 | 1.529 |
| filter=PASS,region,sample(3, AND) | 5.560 | 4.418 | 3.287 | 2.461 | 2.418 | 1.810 | 3.307 | 3.888 | 2.098 | 1.154 | 2.219 | 2.388 |
| filter=PASS,region,sample(3, OR) | 4.430 | 4.218 | 3.401 | 1.914 | 1.485 | 1.481 | 3.148 | 3.339 | 2.068 | 0.923 | 2.251 | 1.945 |
| filter=PASS,region,sample(4, AND) | 4.987 | 5.235 | 4.918 | 2.555 | 2.041 | 1.973 | 4.122 | 4.805 | 2.538 | 1.37 | 3.193 | 2.145 |
| filter=PASS,region,sample(4, OR) | 4.347 | 4.816 | 3.229 | 2.256 | 1.736 | 1.823 | 3.216 | 3.989 | 2.238 | 1.062 | 1.947 | 1.661 |
| filter=PASS,region,sample(5, AND) | 6.695 | 5.556 | 4.779 | 2.783 | 2.996 | 2.452 | 4.835 | 5.793 | 3.087 | 1.714 | 2.779 | 2.553 |
| filter=PASS,region,sample(5, OR) | 5.067 | 5.395 | 3.910 | 2.518 | 2.217 | 1.789 | 4.358 | 4.495 | 2.639 | 1.599 | 2.053 | 1.781 |
| filter=PASS,region=15,sample(3, AND) | 1.894 | 2.714 | 1.539 | 0.463 | 0.841 | 0.460 | 3.070 | 4.768 | 1.535 | 0.904 | 1.716 | 1.616 |
| filter=PASS,region=15,sample(3, OR) | 1.771 | 2.550 | 1.163 | 0.384 | 0.521 | 0.408 | 2.311 | 4.482 | 1.701 | 0.865 | 1.553 | 1.454 |
| Average | 5.487 | 5.126 | 3.628 | 2.115 | 2.412 | 1.816 | 3.750 | 4.573 | 2.218 | 1.180 | 2.206 | 1.947 |
| Speed-up | 1.0 | 1.1 | 1.5 | 2.6 | 2.3 | 3.0 | 1.5 | 1.2 | 2.5 | 4.7 | 2.5 | 2.8 |
| Average – no region 15 | 5.819 | 4.879 | 3.835 | 2.268 | 2.569 | 1.942 | 3.846 | 4.568 | 2.275 | 1.207 | 2.258 | 1.984 |
| Speed-up | 1.0 | 1.2 | 1.5 | 2.6 | 2.3 | 3.0 | 1.5 | 1.3 | 2.6 | 4.8 | 2.6 | 2.9 |
| * Apache Phoenix not working in H | ||||||||||||
GWAS
We defined two cohorts of 100 samples each and run a GWAS analysis over the 208 million variants. The performance observed was very similar to Cohort Stats Calculation. About 2 hours with 10 nodes and about 1 hour with 20 nodes.
Table of Contents: