Getting Genome Browser
To get Jsorolla you can add to your package.json dependency:
Using JavaScript version
First we should define our genome browser. This will contain the general structure of the genome browser and the karyotyope and chromosome panels. To draw these panels, genome browser will use the database "Cellbase" to which also it should be indicated which species we are using. You can see all the species available on Cellbase here.
The target parameter indicates the id of the component wherer the genome browser will be created, in the example there is a "div" called "application". Autorender to draw the only one. If this variable is false, it will be necessary to call the render function of the genome browser. Another parameter of configuration is if we want that the genome browser can be resizable or on the contrary, once defined the size this will be fixed.
| Code Block | ||||||||
|---|---|---|---|---|---|---|---|---|
| ||||||||
let species = {id:"hsapiens", scientificName:"Homo sapiens", assembly:{name:"GRCh37"}};
let region = new Region({chromosome: "13", start: 32996311, end: 33000856}); //initial region
let genomeBrowser = new GenomeBrowser({
client: cellbaseClient,
cellBaseHost: CELLBASE_HOST,
cellBaseVersion: CELLBASE_VERSION,
target: 'application',
width: document.querySelector('#application').getBoundingClientRect().width,
region: region,
species: species,
autoRender: true,
resizable: true,
karyotypePanelConfig: {
collapsed: false,
collapsible: true
},
chromosomePanelConfig: {
collapsed: false,
collapsible: true
},
navigationBarConfig: {
componentsConfig: {
}
},
handlers: {
'region:change': function(e) {
console.log(e)
}
}
}); |
The karyotype and chromosome panels can be passed the configuration if the default is closed or not, or if it is possible to close it or it will always be open.
Line 22-24 are optional, and are useful if you want to add some custom settings to the navigation bar.
Finally we can add to the genome-browser handlers as shown in the example.
At this moment your genome browser would be started and it would look like this:
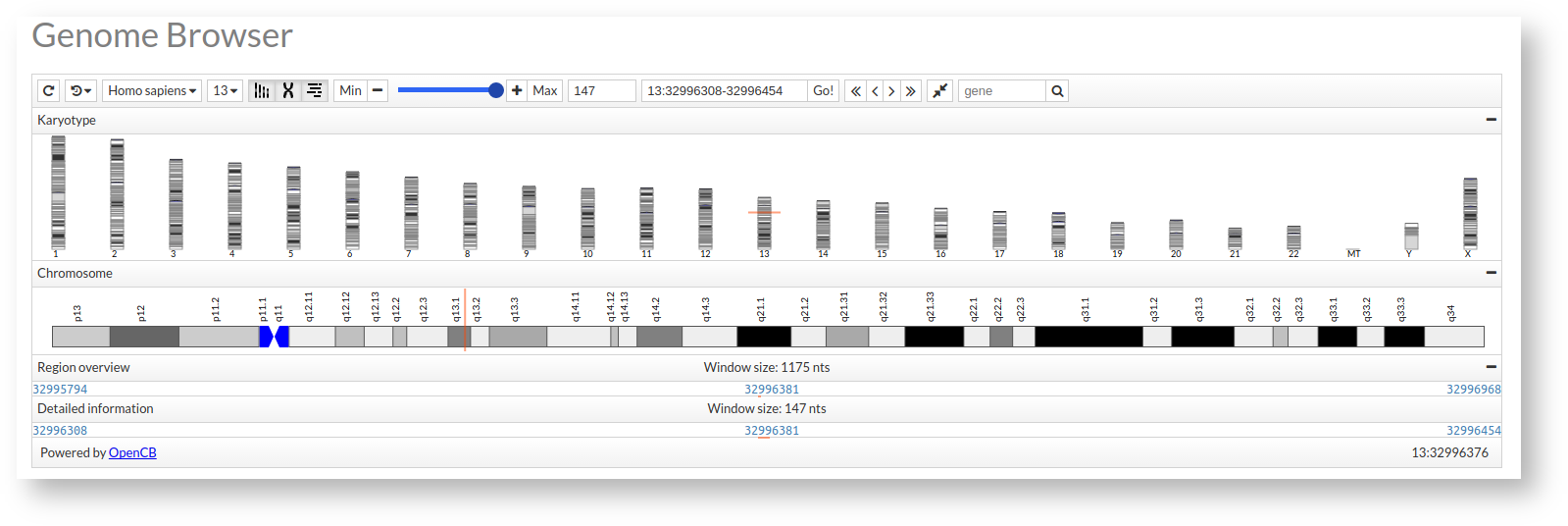
This would be basic form of the genome browser, to enrich it we can define a series of tracks or zones of visualization with different features as explained below.
Overview Track
The particularity of this track is that it has a different zoom to the rest. This smaller zoom (more remote), serves to have an overview of what is in that area soight while in the rest of tracks you see in more detail the zone.
We will define a FeatureTrack to which we will pass the renderer, the adapter and other parameters of configuration of the track like the title, the minimun size where it changes histogram(minHistogramRegionSize), the maximun size where the names of the components display (maxLabelRegionSize) or height.
FeatureRenderer is the renderer in charge of painting a boz for each feature. Adding the gene type (FEATURE_TYPES.gene) knows how to draw the color fo each feature and the tag among other things.
The data will be collected with a CellBaseAdapter, indicating that we want to collect the genes and exclude the transcripts since being a distant zoom if we painted them woild be difficult to visualize.
| Code Block | ||||||
|---|---|---|---|---|---|---|
| ||||||
let gene = new FeatureTrack({
title: 'Gene overview',
minHistogramRegionSize: 20000000,
maxLabelRegionSize: 10000000,
height: 80,
renderer: new FeatureRenderer(FEATURE_TYPES.gene),
dataAdapter: new CellBaseAdapter(cellbaseClient, "genomic", "region", "gene", {
exclude: 'transcripts,chunkIds'
}, {
chunkSize: 100000
})
});
genomeBrowser.addOverviewTrack(gene); |
Sequence Track
The sequence track shows that nucleotide matches in the position we are looking for.
This track accpets in this definition the title, the height of the track, the maximum size of nucletotics in which the track continues painting(visibleRegionSize), the renderer and the CellBaseAdapter. We recommend that the value of the visibleRegion not be very high because the sequence would be clearly seen when painting thousand of letters.
| Code Block | ||||||||
|---|---|---|---|---|---|---|---|---|
| ||||||||
let tracks = [];
this.sequence = new FeatureTrack({
title: 'Sequence',
height: 20,
visibleRegionSize: 200,
renderer: new SequenceRenderer(),
dataAdapter: new CellBaseAdapter(cellbaseClient, "genomic", "region", "sequence", {}, { chunkSize: 100})
});
tracks.push(this.sequence);
genomeBrowser.addTrack(tracks); |
To add this track to the genome browser, we will define an array of tracks that we will add all the track we want to add (except overviewtrack), and later add this to the genome browser as it does on line 10.
Gene Track
Adding a gene track is similar to making a sequence track since we will also define it and add to the track array which we then add to the genomeBrowser.
Gene track shows the genes and transcripts that are in the region visualized, for this we consult the cellbase database to which we pass which species we are consulting.
| Code Block | ||||
|---|---|---|---|---|
| ||||
this.gene = new GeneTrack({
title: 'Gene',
minHistogramRegionSize: 20000000,
maxLabelRegionSize: 10000000,
minTranscriptRegionSize: 200000,
height: 120,
cellbase: {
"host": CELLBASE_HOST,
"version": CELLBASE_VERSION,
"species": "hsapiens"
}
});
tracks.push(this.gene); |
If we want to change the renderer or defaunt adapter of the gene track we can indicate it with the parameters renderer and adapter respectively.
| Code Block | ||||
|---|---|---|---|---|
| ||||
this.gene = new GeneTrack({
title: 'Gene',
minHistogramRegionSize: 20000000,
maxLabelRegionSize: 10000000,
minTranscriptRegionSize: 200000,
height: 120,
cellbase: {
"host": CELLBASE_HOST,
"version": CELLBASE_VERSION,
"species": "hsapiens"
}
renderer: new GeneRenderer({
handlers: {
'feature:click': function(e) {
console.log(e)
}
}
})
dataAdapter: new CellBaseAdapter(cellbaseClient, "genomic", "region", "gene", {
exclude: 'transcripts.tfbs,transcripts.xrefs,transcripts.exons.sequence'
}, {
chunkSize: 100000
})
});
tracks.push(this.gene); |
Variant Track
The variant track has two operating modes: individual or family. If we pass name of one of ours will understand that these samples correspond to a family and will activate the family mode, otherwise the individual mode will be activated.
Individual mode:
In the individual mode each variant is and individual and so is shown. In the example as they are variants of type snp, we pass the configuration of the painting that we want to the renderer. This configuration is defined in the config file of the source code, inside the genome-browser.js then the library is downloaded with npm.
| Code Block | ||||
|---|---|---|---|---|
| ||||
let variant = new VariantTrack({
title: "Variant",
closable: true,
minHistogramRegionSize: 20000000,
maxLabelRegionSize: 10000000,
minTranscriptRegionSize: 200000,
visibleRegionSize: 100000000,
height: 300,
opencga: {
client: opencgaClient,
studies: "platinum:illumina_platinum"
}
});
tracks.push(variant); |
Family mode:
When we define the samples in the variant track the family mode is activated. This shows in the left side the name of the sample and in horizontal we can see the variants of each sample.
| Code Block | ||||
|---|---|---|---|---|
| ||||
let variant = new VariantTrack({
title: "Variant for family",
closable: true,
minHistogramRegionSize: 20000000,
maxLabelRegionSize: 10000000,
minTranscriptRegionSize: 200000,
visibleRegionSize: 100000000,
height: 300,
opencga: {
client: opencgaClient,
studies: "reference_grch37:1kG_phase3",
samples: ["HG00096", "HG00097", "HG00099"]
}
});
tracks.push(variant); |
When you use the opencga parameter, the genome browser itself understands that the adapter we want to user is the OpencgaAdapter. The renderer to use is the one that comes by default defined in the variant track. This renderer is the FeatureRender using the FeatureType.Variant configuration. This FeatuareType, there in config archive in the code or genome-browser.js when you getting jsorolla using npm.
The code equivalent to the above but using the adapter definition and renderer would be as follows:
| Code Block | ||||
|---|---|---|---|---|
| ||||
let variant = new VariantTrack({
title: "Variant",
closable: true,
minHistogramRegionSize: 20000000,
maxLabelRegionSize: 10000000,
minTranscriptRegionSize: 200000,
visibleRegionSize: 100000000,
height: 300,
renderer: new FeatureRenderer(FEATURE_TYPES.variant),
dataAdapter: new OpencgaAdapter(opencgaClient, "analysis/variant", undefined, "query",
{
studies: "platinum:illumina_platinum",
// exclude: "studies.files,studies.stats,annotation" //Exclude for samples mode
exclude: "studies,annotation" //Exclude for individual variation mode
}, {
chunkSize: 100000
})
});
tracks.push(variant); |
It is important for the developer to understand that the genome browser always uses a track, an adapter, and a renderer even though it does not look the same in its definition. If the developer wants to change the renderer or the adapter using the default tracks should include the adapter or renderer they want in the definition of the track.
Aligment Track
The aligment track is made up at the top of the coverage and at he bottom of the bams reads.
| Code Block | ||||
|---|---|---|---|---|
| ||||
let alignment = new AlignmentTrack({
title: "Alignment",
closable: true,
minHistogramRegionSize: 5000,
maxLabelRegionSize: 3000,
visibleRegionSize: 100000000,
height: 300,
renderer: new AlignmentRenderer(FEATURE_TYPES.bam),
dataAdapter: new OpencgaAdapter(opencgaClient, "analysis/alignment", undefined, "query",
{
fileId: fileId,
study: study
})
});
tracks.push(alignment); |
When you use the adapter of Opencga to visualize the bams you must pass the name of the files to him by the parameter fileId.
Custom Track
You may want to collect data from another database but you don't want to implement a custom adapter for yourself. The solution to this problem is to use the adapter template.
In the following example, instead we create a variant track using a template adapter instead of our adapter for the cellbase database.
| Code Block | ||||
|---|---|---|---|---|
| ||||
let t = new FeatureTrack({
title: 'Variant',
closable: true,
minHistogramRegionSize: 20000000,
maxLabelRegionSize: 10000000,
minTranscriptRegionSize: 200000,
histogramColor: '#92de47',
height: 100,
renderer: new FeatureRenderer(FEATURE_TYPES.snp),
dataAdapter: new FeatureTemplateAdapter({
multiRegions: false,
histogramMultiRegions: false,
uriTemplate:"http://ws.babelomics.org/cellbase/webservices/rest/v4/hsapiens/genomic/region/13:32990000-32999999/snp? exclude={exclude}",
templateVariables: {
exclude: 'annotation.populationFrequencies,annotation.additionalAttributes,transcriptVariations,xrefs,samples'
},
species: genomeBrowser.species,
cacheConfig: {
chunkSize: 100000
},
parse: function(response) {
var chunks = [];
for(var i = 0; i< response.response.length; i++){
var res = response.response[i].result;
chunks.push(res);
}
return chunks;
}
})
});
tracks.push(t); |
| Param | Type | Description |
|---|---|---|
| multiRegions | boolean | Indicates if large regions will make a single call to the server or several. |
| histogramMultiRegions | boolean | Indicates whether large regions will make a single call to the server or multiple calls when the display zone is in histogram mode. |
| uriTemplate | string | uri of the server to which calls will be made. The value of the query params will be indicated by a "{variable}". This value will be declared in templateVariables. |
| templateVariables | object | Object that includes the value of each query param of the uriTemplate. As query param will be defined as: in the key, the name of the query params that we have put in the uriTemplate and as value, the value to be replaced in the uriTemplate. |
| species | ||
| cacheConfig | object | |
| parse | function |
__________________________________________________________________________________________________________________
Using Polymer web component
To facilitate the use of the Genome Browser component, a WebComponent has been created that you can import into your html page.
In this tutorial you will learn how use this component for visualizate differents tracks.
In first time, you must import the component in your page.
| Code Block | ||||||
|---|---|---|---|---|---|---|
| ||||||
<link rel="import" href="jsorolla/dist/core/webcomponent/genome-browser.html"> |
Subsequently, we will declare the genome component browser in our html code. For this we must know the list of parameters that the component accepts.
Here is a list of parameters that the component accepts and uses:
| Param | Type | Description |
|---|---|---|
| cellbase-client | Object | Client for use Cellbase. Necessary for: Gene track (using cellbase adapter, default). |
| opencga-client | Object | Client for use opecngaDB. Necessary for: Varriant Track and Aligment Track (using opencga adapter, default). |
| project | Object | To use opencga webservices, you will need to know which project the data to display. |
| study | Object | To use the opencga webservices, this will need to know in which study within a project, are the data to be visualized. |
| region | Object | Region where the genome-maps will initially be positioned. |
| species | Object | Species to draw. Default: homo sapiens. |
| width | Number | Component width. |
| active | Boolean | Enables or disables the component. If it is disabled the component will not be displayed.Default: false. |
| settings | Object | This parameter sets other settings of the genome maps that the user wants to pass. |
Finally, declare the component. In this case, we only defined the GeneTrack and Sequence track:
| Code Block | ||||
|---|---|---|---|---|
| ||||
<genome-browser cellbase-client="{{cellbaseClient}}" region="{{region}}" tracks="{{tracks}}" active="true">
</genome-browser>
|
In the javascript part we will give value to the variables:
| Code Block | ||||
|---|---|---|---|---|
| ||||
CELLBASE_HOST = "bioinfo.hpc.cam.ac.uk/cellbase";
CELLBASE_VERSION = "v4";
let cellBaseClientConfig = new CellBaseClientConfig(CELLBASE_HOST, CELLBASE_VERSION);
this.cellbaseClient = new CellBaseClient(cellBaseClientConfig);
this.region = new Region({chromosome: "11", start: 68177378, end: 68177510});
this.tracks ={sequence: {type: "sequence"}, gene: {type: "gene"}};
|
We have created a cellbase client that will connecto to the Host "bioinfo.hpc.cam.ac.uk/Cellbase". If we do not want to change the installation of Cellbase, it is not necessary that we define the host and the version, but we initialize a client. With the parameter "region", we ask that the display is at "11:68177378-68177510". It's possible to not see these positions in the display where the painted region is shown, but you well see a window frame containing the region. This is because the genome browser adjusts the display to optimize the cache.
Finally, the last param is "tracks", in this case we want see the track "sequence" and "gene". We can define other types of tracks as follows:
- Individual Variant Track (for example SNP): {trackname: {type: "variant", config: {} }}.
- Family Variant Track: {trackname: {type:"variant", config:{samples:"sample1","sample2"}}}.
- Aligment Track:{trackname:{type:"alignment", config:{files:"bam1", "bam2"}}}.
Table of Contents:
| Table of Contents | ||
|---|---|---|
|