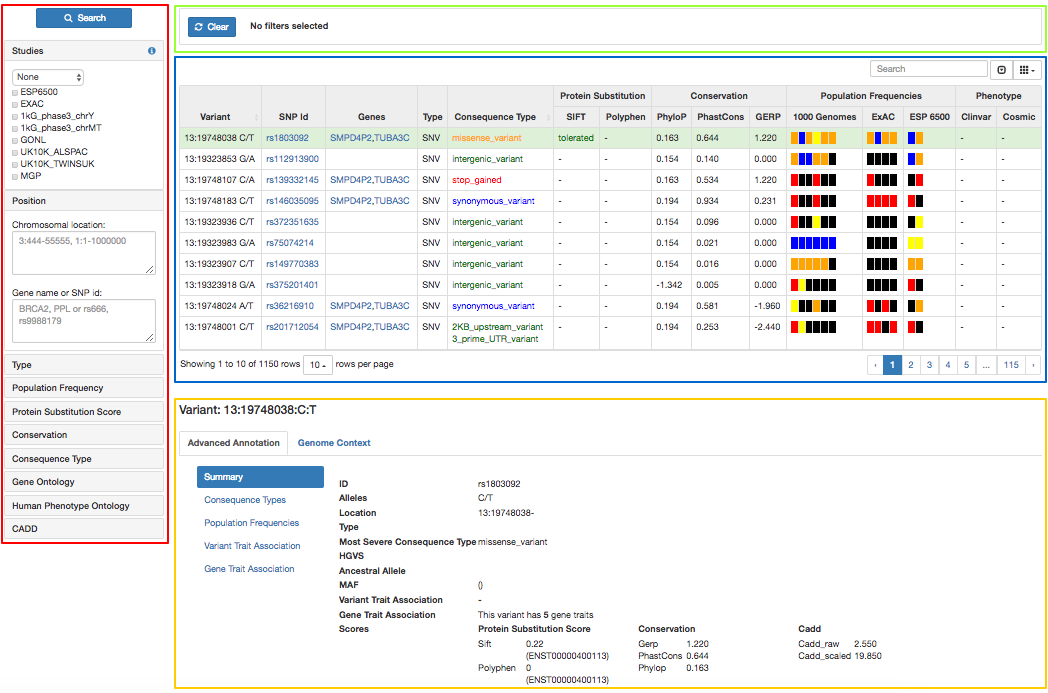
- Filter panel (red): Contains the list of available parameters to filter the list of variants in the main panel
- Active filters (green): List of applied filters
- Main panel (blue): Table containing the list of variants and extra information about them
- Variant detail (yellow): Contains specific and extended information about the variant selected in the main panel
Filter panel
The list of variants in the main panel can be thoroughly filtered by any of the parameters listed below. Some sections appear collapsed by default, to expand them, just click on the title.
- Studies: users can filter the list of variants by its existence or absence in one or more published studies. To do so, select the studies to use and the desired option from the dropdown menu: Only in to include variants that appear only in all the selected studies but not in the unselected ones, In to include variants that appear in any of the selected studies but not in the unselected ones, At least in to include variants that appear in any of the selected studies and can also be present in the unselected ones, and, Not in to exclude all the variants that appear in any of the selected projects.
- Position: restrict the list of variants to a specified genomic region (specified by chr:start-end), gene (in HGNC symbol) or SNP (use the rsID).
- Type: Limit the search to just one or few types of variant: SNV (Single Nucleotide Variant), MNV (Multi-nucleotide Variant), CNV (Copy Number Variant), SV (Structural Variant) or INDEL (Insertion or deletion).
- Population Frequency: restrict the list of variants to those which have a MAF (Minor Allele Frequency) higher/lower/equal to a defined value in the 1000 genomes project, ExAc and/or ESP 6500. To do so, expand the "+" sign next to the desired project, select from the dropdown menu the comparison operator and specify the value (between 0 and 1) in the text box. MAF filtering can be applied to all of the samples in the specified project (ALL) or to one/few specific populations within the selected project.
- Protein Substitution Score: users can also filter by the annotated protein substitution scores such as SIFT and PolyPhen. Filtering can be done using their prediction keywords from the dropdown list or using an specified score. For the latter, just select "Score..." in the list of options, chose the comparison operator and specify a value (between 0 and 1) in the text box. Bear in mind that a score close to 0 in SIFT is considered deleterious while a score close to 0 in PolyPhen is considered benign.
- Conservation: filter by conservation scores using PhyloP, PhastCons and/or Gerp.
- Consequence Type: restrict the list of variants to those annotated with a particular consequence type.
- Gene Ontology: limit the list of variants only to the ones in a gene annotated with the specified Gene Ontology term(s).
- Human Phenotype Ontology: limit the list of variants only to the ones in a gene annotated with the specified Human Phenotype Ontology term(s).
- CADD: filter the search using CADD score. Users can chose to filter by the "raw" score or the "scaled" score. Please, see CADD's website to find information about the two options: http://cadd.gs.washington.edu/info
After one or more filters have been set, just click on the Search button at the top of the panel to apply them.
Active filters
Every time a filter is applied, this panel refreshes and includes a button with the filter. Users can easily remove individual filters just by clicking on the buttons in the panel. To remove all the active filters click on "Clear" at the left hand side of the panel.
Main panel
The main panel contains a table with the list of variants from a specified project/study. This list can be modified by any of the parameters in the filter panel. The table shows one row per variant and several columns containing general information about each variant:
- Variant: the variant coded as chr:position ref/alt
- SNP Id: if the variant is included in dbSNP, this field will show the rsID, otherwise, it will be empty
- Genes: if the variant overlaps with a gene, the gene symbol will be displayed here, otherwise, it will be empty. If it overlaps with more than one gene, they will be listed separated by comma
- Type: type of variant, SNV (Single Nucleotide Variant), MNV (Multi-nucleotide Variant), CNV (Copy Number Variant), SV (Structural Variant) or INDEL (Insertion or deletion)
- Consequence Type: this is the predicted effect of the variant in the genome. If a variant has more than one consequence type (because it overlaps different transcripts and/or genes), the "worst" consequence will be displayed. Consequence types are also colour coded according to their impact to improve readability:
- High impact in red
- Moderate impact in orange
- Low impact in blue
- Modifier in green
Deleteriousness: It consists of three sub-columns - SIFT, Polyphen and CADD.
SIFT scores are classified into tolerated and deleterious. SIFT score takes values in the range [0, infinite[, the lower the values, the more damaging the prediction. Color code for SIFT predictions: Tolerated in green and Deleterious in red.
Polyphen scores are classified into benign, possibly damaging, probably damaging and possibly & probably damaging. To visualize the actual score value, Please leave the cursor over each tag . Polyphen score takes values in the range [0, 1[, the closer to 2, the more damaging the prediction. Color codes for polyphen predictions:
Probably damaging in red
Possibly damaging in orange
Benign in green
Unknown in black
The CADD column displays the the scaled CADD score. A scaled C-score of greater of equal 10 indicates that the substitution is predicted to be within the 10% most deleterious substitutions that you can do to the human genome, a score of greater or equal 20 indicates the 1% most deleterious and so on. If you would like to apply a cut-off on deleteriousness, it is suggested by the CADD authors to put a cutoff somewhere between 10 and 20. Maybe at 15, as this also happens to be the median value for all possible canonical splice site changes and non-synonymous variants. Scores equal or greater than 15 are displayed in red.
Conservation: It consists of three columns - PhyloP, PhastCons and GERP.
Positive PhyloP scores measure conservation which is slower evolution than expected, at sites that are predicted to be conserved. Negative PhyloP scores measure acceleration, which is faster evolution than expected, at sites that are predicted to be fast-evolving. Absolute values of phyloP scores represent -log p-values under a null hypothesis of neutral evolution.
PhastCons estimates the probability that each nucleotide belongs to a conserved element. The phastCons scores represent probabilities of negative selection and range between 0 and 1.
Positive GERP scores represent a substitution deficit (i.e., fewer substitutions than the average neutral site) and thus indicate that a site may be under evolutionary constraint. Negative scores indicate that a site is probably evolving neutrally. Some authors suggest that a score threshold of 2 provides high sensitivity while still strongly enriching for truly constrained sites.
- Population Frequency: Minor Allele Frequency (MAF) are shown for the given populations and studies in the config.js. According to the frequency range, they are classified as follows:
- Very Rare if MAF is less than 0.001 and it is displayed in bright red.
- Rare if MAF ranges from 0.001 to 0.005 and it is displayed in pale red.
- Average if MAF ranges from 0.005 to 0.05 and it is shown in light blue.
- Common if the MAF is greater than 0.05 and it is shown in dark blue.
- Phenotype: It displays the traits observed for the variants. The sources taken are ClinVar and Cosmic.
Variant detail
By default, the first variant in the grid is selected and the tabs are loaded with information for that variant.
The tabs are
- Advanced Annotation: Here, the detailed annotation of the variant can be seen. Details like consequence types, population frequencies, variant trait association and gene trait association are included.
- Genotype Stats: The genotype frequencies and counts from various cohorts from each study are tabulated under this tab.
- Genome Context: This tab includes the genomic context viewer.
- Beacon Network: This network contains a list of hosts to query. These hosts are configured in config.js under beacon section. When the search button is clicked, it searches over the provided hosts and displays the results in a table.
Table of Contents: